APPENDIX
Table of Contents
Topics Page Number
Distillation A-1
Reflux A-5
Liquid-Liquid Extraction A-6
Recrystallization A-7
Gas Chromatography A-9
Infrared Analysis A-10
Distillation
When pure water is heated in the distillation apparatus shown in Figure l, there is an increased tendency for molecules to escape from the surface of the liquid (distilland). Thus, the vapor pressure of the liquid increases until it becomes equal to the atmospheric pressure and the liquid begins to boil. Continued heating supplies the heat of vaporization necessary for further conversion of liquid to gas. Thus, vapors rise, warm the stillhead, and begin flowing into the condenser, which is cooled by water. Vapors passing through the condenser are therefore cooled to give a liquid condensate, the distillate, which can be collected, in a receiving flask.
Distillation should be done steadily and at such a rate that the thermometer bulb always carries a drop of condensate and is bathed in a flow of vapor. Liquid and vapor are then in equilibrium around the bulb, and the temperature registered is the true boiling point of the liquid. If excessive heat is applied, the vapor becomes superheated, the drop disappears, the liquid-vapor equilibrium is upset, and the temperature rises above the boiling point.
Since all of the heat being supplied isn't immediately dissipated by vaporization, some superheating of the liquid may occur. A thermometer immersed in the boiling liquid would therefore record a temperature a little above the boiling point, but a thermometer in the vapor space shown in Figure l records the true boiling point, even if the liquid is superheated or if it contains a nonvolatile solvent. For example, when a solution of sugar in water is distilled, the boiling point recorded on a thermometer in the vapor phase is 100 degrees (at 760 mm) throughout the distillation. Whereas the temperature of the boiling liquid is initially somewhat above 100 degrees and continues to rise as the sugar solution becomes more concentrated. The vapor pressure of the sugar solution is dependent upon the number of water and sugar molecules present in a given volume. Hence, the vapor pressure at any given temperature decreases with increasing concentration of nonvolatile sugar molecules and decreasing concentration of water, and a higher temperature is required for boiling. However, sugar molecules do not leave the solution, and the drop clinging to the thermometer bulb is pure water in equilibrium with pure water vapor.
When a distillation is done in a system open to air, the boiling point is the temperature at which the pressure of the boiling liquid equals that of the atmosphere. The prevailing
barometric pressure should be noted and allowance should be made
Figures 1-4: Distillation Setups
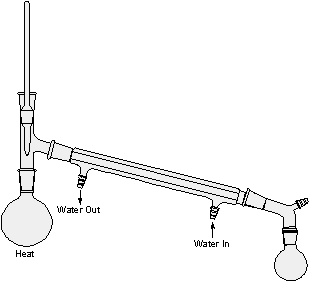
figure 1: Simple Distillation
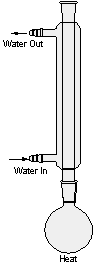
figure 2: Reflux
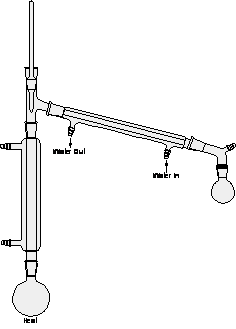
figure 3: Fractional Distillation
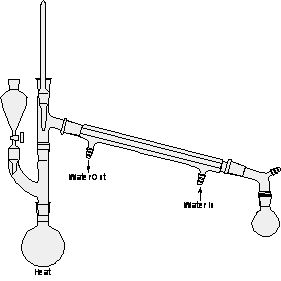
figure 4: Concentration of a Solvent
Note: Be sure to support or clamp all round bottom flasks and grease all joints. Make sure all joints are sealed and that cooling water goes in the lower entrance of the condenser and exits the upper entrance. Secure the exit hose in a cup sink to prevent floods in the lab. In figures 1-4 all the condensers are water cooled except the vertical one in figure 3, the fractionating column. For appreciable deviations from the normal pressure of 760 mm by reference to a table like Table I, distillation can also be done under a vacuum created by an oil or water pump with substantial reduction of boiling point.
TABLE I VARIATION IN BOILING POINT WITH PRESSURE
Pressure Boiling Point
Water Benzene
780 mm 100.7oC 81.2oC
770 mm 100.4oC 80.3oC
760 mm 100.0oC 80.1oC
750 mm` 99.6oC 79.9oC
740 mm 99.3oC 79.5oC
584 mm* 92.8oC 71.2oC
*Institute de Quimica, Mexico City, altitude 7700 ft(2310 meters)
Mixtures of the volatile and miscible liquids carbon tetrachloride (b. pt. 76.8 degrees C) and toluene (b.p. 100.6 degrees C) distill at temperatures intermediate between the two boiling points. The composition of the distillates changes progressively during the distillation process. Since the two substances are mutually soluble, each is diluted by the other and there is a consequent reduction in the vapor pressure exerted by the mixture at any given temperature. The mixture, however, exerts a common pressure against the atmosphere and boiling occurs when the sum of the two partial pressures of the liquids equals atmospheric pressure.
An azeotropic solution is a mixture of liquids of a certain definite composition that distills at a constant temperature without change in composition. The boiling point is usually lower than that of the lowest boiling component, but it is sometimes higher than that of the highest boiling component; it is never in an intermediate range. Ordinary ethyl alcohol is an azeotrope of b.p. 78.1 degrees C composed of 95.5 % ethanol (b.p. 78.4 degrees C) and 4.5% water (b.p. 100.0 degrees C) by weight. An azeotropic solution of 32.4% ethanol and 67.6% benzene (b.p. 80.1 degrees C) boils at 68.2 degrees. A ternary azeotrope containing 74.1% benzene, 18.5% ethanol, and 7.4% water boils at 64.2 degrees C. Adding benzene to 95% alcohol and removing the water in the volatile azeotrope makes absolute ethanol.
Fractional Distillation
Methanol has a boiling point (64.7 degrees C) about 35 degrees below the boiling point of water, with which it is miscible. When a mixture of equal volumes of the two liquids is distilled using the sample apparatus of Figure l, the initial boiling point is a little above that of methanol and the final boiling point a little below that of water. All fractions of the distillate are found to be mixtures, and little separation of the two components is achieved. Use of an efficient fractionating column markedly alters the situation and permits sharp separation of the two liquids. The fractionating column shown in the assembly of Figure 3 contains a section of stainless steel scouring sponge, which forms a porous packing for equilibration of vapor and condensate. After the methanol-water mixture has been boiled until rising vapor has warmed the column over the entire length, a less volatile part of the vapor condenses and trickles down through the crevices in the packing. Fresh vapor from the flask forces its way up through the descending condensate with attendant heat interchange, since the vapor is hotter than the trickling condensate liquid. A more volatile part of the liquid vaporizes and a less volatile part of the vapor condenses. Equilibrations of this type occur in all parts of the column. The vapor that eventually passes into the receiver is highly enriched in the more volatile component, whereas the condensate that continually drops back into the flask is scrubbed of the volatile liquid and enriched in the less volatile one.
Using the column illustrated in Figure 3, it is possible to effect a sharp separation of a mixture of liquids boiling only 35 degrees apart; see Figure 5. To obtain so successful a result, however, one must use a very uniform, constant heat source and boil the liquid slowly and steadily enough to allow full heat equilibration between liquid and vapor in the column. As the distillation of methanol and water progresses, we can see from Figure 5 that the composition of the distillate changes. The methanol will be the first to be collected since it has a lower boiling point. The temperature remains constant at the boiling point of methanol while it is distilled. Next the temperature rapidly increases to that of water while little distillate is collected. The collection flask would normally be changed during any rapid increase in temperature such that different fractions or samples are taken. Once the boiling point of water is reached, the temperature remains constant again while water is distilled and collected. Column efficiency is improved by insulating the column, packing the column with steel wool, and by introducing a small condenser at the very top of the column to effect partial condensation of the already highly rectified vapor. A fractional distillation apparatus permits observation of the functioning of a column and will demonstrate an enormous efficiency over simple distillation.
TABLE II
Properties of Solvents
Solvent Boiling Pt Latent Heat of Surface
(oC) Vaporization Tension
(cal/gram) (20oC)
(dynes/cm)
Acetone 56.5 125.3 23.7
Methanol 64.7 216.7 22.6
Hexane 68.7 79.2 18.4
Carbon
Tetrachloride 76.7 46.4 26.8
Benzene 80.l 93.5 29.0
Water 100.0 536.6 72.7
Toluene 110.6 86.8 28.4
Xylene 137-144 93.4 28.9
Since the separation effected in a fractionating column is dependent upon heat equilibration in multiple processes of vaporization and condensation, efficiency of separation increases with increasing differences in the heats of vaporization of the liquids concerned. Table II shows that the heat of vaporization of water far surpasses that of any of the organic solvents listed. Thus, water is a particularly favorable component for separation from a mixture. Water also surpasses organic liquids in surface tension, a property that determines the behavior of a condensate on a packing surface. The high heat of vaporization, the high surface tension, and a boiling point that is high for a substance of so low molecular weight (compare water with ammonia) are all consequences of a high degree of association of molecules in liquid water.
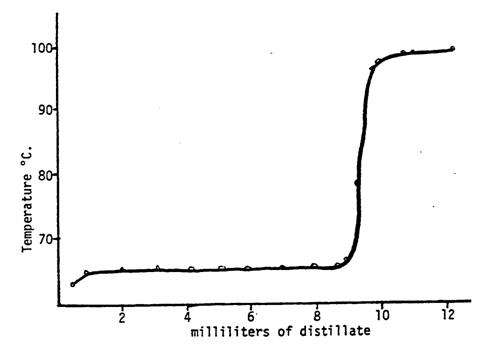
Figure 5 Fractionation of a mixture of methanol (b.p. 64.7oC) and water.
Steam Distillation
As discussed above, the boiling point is the temperature at which the total vapor pressure of a mixture equals atmospheric pressure. When two immiscible liquids (liquids which have very low solubility in each other) are heated, each exerts its own vapor pressure independently of the other. Consequently, the boiling point of the mixture is much lower than it would be for either component or for a mixture of two liquids that are miscible.
This property is used in the process of STEAM DISTILLATION, in which a compound that is virtually immiscible with water can be distilled at a temperature much lower than its normal boiling point. This has the added advantage that there will be less decomposition of the compound because of the lower temperature.
Reflux
Heating accelerates the rate of a chemical reaction by increasing the average kinetic energy of the molecules, so that a larger fraction of molecules will have sufficient energy to react at a given instant. Using a reaction solvent that has a boiling point within the desired temperature range for the reaction can most easily control the temperature of a chemical reaction. The reaction is carried out at the boiling point of the solvent, using a water-cooled condenser to return the vapors to the reaction vessel and prevent their escape. This is the essence of reflux. It is the most widely used technique for carrying out organic reactions at elevated temperatures.
Liquid-Liquid Extraction
Liquid-liquid extraction is one of the many purification or separation techniques used in organic chemistry in addition to distillation and recrystallization. Paper, thin film, column, gas, and high performance liquid chromatography are all separation techniques related to liquid-liquid extraction in that components are separated based on their relative solubility in two different phases (immiscible solvents). As a general rule, polar compounds tend to dissolve in polar solvents, and nonpolar compounds tend to dissolve in nonpolar solvents; or more succinctly "like dissolves like." Solubility is also a temperature dependent property-most chemical can be dissolved if enough heat is applied. We will often cool a mixture to make something less soluble to improve the recovery and yield. We will use these general principles to separate compounds in this course.
We will use liquid-liquid extraction or washing to take advantage of the differences in solubility of the components to purify and separate compounds. In liquid-liquid extraction we bring two immiscible (completely mutually insoluble) liquids in contact. These are usually an organic liquid (organic solvent) and water. If they are not in motion, two separate distinct layers will form, the bottom one being the liquid with the greater density. We can increase the contact between the two liquids, however, by shaking them in a separatory funnel. Care should be take to make sure the stopcock is closed before filling the separatory funnel. The funnel should also be inverted and vented several times during the shaking process to release pressure if volatile liquids are used. Be sure to hold the stopper firmly in place at all times when shaking. Practice using the separatory funnel with water if you have never used one before.
Suppose that an organic solution containing a neutral nonpolar organic compound and a carboxylic acid is shaken with a dilute aqueous sodium hydroxide solution and then allowed to stand until the two layers separate. During the shaking process, the hydroxide ion will react with only the carboxylic acid component of the mixture to form the water soluble carboxylate anion. This changes the solubility properties of the acid and results in most of it moving from the organic liquid layer to hydroxide ion-water layer. The carboxylate ion thus is called a solute and is said to have been extracted into the aqueous (water) phase. The two phases are then separated into two fractions: The aqueous sodium hydroxide solution containing the carboxylate anion as its salt and the organic layer containing the neutral compound.
The two phases will separate and form two separate layers based on differences in polarity and density. The organic layer is much less polar and has a much lower density compared to the diluted NaOH solution. Sometimes the difference in polarity and/or density of the two phases may not be great enough to effect a separation causing the formation of an emulsion. The separation can sometime be improved by adding more of each solution or solvent.
It is important to note that single extractions do not necessarily yield complete separations, and that multiple extractions may be needed. In experiment 7, you will extract the original organic solution two times with aqueous sodium hydroxide solution to remove the acid and water-soluble impurities from the organic layer. The two aqueous extracts are then combined and set aside as the aqueous sodium hydroxide fraction. The organic solution is further extracted once with distilled water to remove any water-soluble impurities. Once these extractions are complete, the organic solution should contain only the "neutral" compound. Several other experiments will purify products using liquid-liquid extraction or washing process.
Drying Agents
Organic liquids and solutions that have been separated from a reaction mixture or isolated from a natural product often retain traces of water. Most drying agents are inorganic salts that form hydrates by combining chemically with water:
Na2SO4 + nH2O ® Na2SO4·nH2O
So how much drying agent do you add to remove the water? As a general rule: one gram of drying agent per 25 mL of liquid. Carefully observe while adding enough drying agent that it does not clump upon addition. If it is clumping, it is not complexing because there is no water to complex with!
Recrystallization
The principle of recrystallization is quite simple (refer to figure 6 on the next page). The solid to be purified is redissolved in an appropriate solvent and allowed to reprecipitate or recrystallize more slowly thus preventing the entrapment of impurities in the crystal lattice.
The solubility of a solute in a solvent is a function not only of the chemical structure of the two components, but also of the temperature. Do you remember that temperature is a measure of a molecule’s kinetic energy? Ordinarily the solubility of a solid in a liquid increases with temperature, and, in some instances, the effect may be very marked (Table below). This phenomenon is of great importance to the chemist because it is the basis for the purification of solids by recrystallization.
SOLUBILITY OF SUCCINIC ACID WITH TEMPERATURE
Solvent Temperature Solubility
oC g/100 ml
Water 20 7
Water 100 121
If a solid is dissolved in a hot solvent and the solution is cooled, the saturation point may be reached. Any subsequent cooling of the solvent results in the separation of solid by recrystallization. If the original material contains any foreign matter, a highly specific purification procedure using an appropriate solvent can be devised. Contaminants that are insoluble in the solvent can be separated by filtration of the hot solution; small amounts of those that are highly soluble in the solvent will remain in solution as the desired product crystals are formed and removed by filtration. Occasionally it is necessary to remove traces of highly colored impurities by boiling the solution with decolorizing charcoal (on which the impurities absorb) removing the charcoal by filtration, and proceeding with the crystallization.
Figure 6 – Steps in Recrystallization
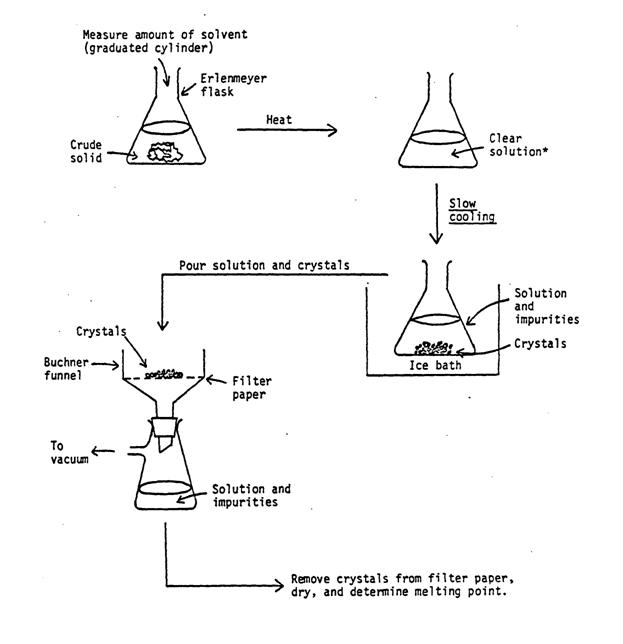
* If solids (sand, dirt, etc.) remain at this step, filter the hot solution to remove any insoluble left in the flask.
Many times the most efficient solvent for recrystallization is a mixture of two liquids. Such mixed solvents are used when a solid is too soluble in one component and too insoluble in the other for convenient crystallization. The material to be crystallized is usually dissolved in the hot solvent in which it is highly soluble, then the other solvent is added slowly to the hot solution until the solute tends to separate (the solution will turn cloudy). The mixture is then heated again to bring all the materials back into solution (if necessary, small amounts of the first
solvent are added to aid the process). Slow cooling will result in separation of crystalline product. Water-methanol or water-ethanol are common solvent pairs that find general use.
Vacuum or Suction Filtration
The pure crystals generated by recrystallization can be collected by vacuum filtration. In this process a Buchner funnel is in a filter flask with a rubber adapter fitting between the two. (A Hirsch funnel can be used for samples smaller than a gram.) A circle of filter paper is placed inside the funnel. The paper should be big enough to cover the holes in the funnel but small enough to lay flat. The filter flask should be connected to a source of vacuum with thick wall tubing (if the tubing collapses under vacuum you’ve got the wrong tubing!). A trap should be placed between the filter flask and the source of vacuum if there is any chance of vapor or liquid being drawn into the vacuum line. A clamp and ringstand can be used to secure the flask so that it will not fall over.
Once the vacuum filtration setup is assembled add a small amount of the solvent or supernate to moisten the filter paper. Turn on the vacuum to seal the paper against the funnel and pour the material to be filter into the funnel evenly. Make sure the filter flask is never more than half full. If this occurs turn off the vacuum and pour out the filtrate into another flask. This filtrate can also be used to rinse any remaining crystals into the funnel. After removing the filtrate, the cold wash liquid can be poured into the funnel with the vacuum off. After 1-2 minutes turn on the vacuum and leave it on for several minutes to dry the crystals.
Gas Chromatography
Gas chromatography is a good method for determining the purity of a volatile liquid organic compound such as cyclohexene. Gas chromatography separates compounds based on physical properties such as polarity and molecular weight. The separation and detection of the components of a sample are accomplished in short by the following process (see figure 7).
Figure 7 – Components of a Gas Chromatograph
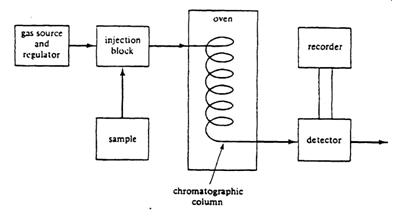
The sample is injected into one end of a heated column which contains a stationary phase and through which a carrier gas such as helium is flowing. The sample vaporizes and is transported into and through the column by the carrier gas. As the sample travels through the column, the components are separated based on their partial solubility in the stationary phase such that the least soluble component will exit the column first and the most soluble component last. The separated components will then travel through a detector that generates a gas chromatogram (see Figure 8) via an electrical signal to a recorder. The peaks in the chromatogram are a result of various components of the sample passing through the detector. The components are characterized by their respective retention times. In a one component sample only an air peak and the peak for the component would be noted. If there are impurities in the sample there will be additional peaks.
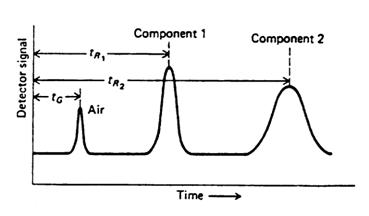
Figure 8 - Gas Chromatogram For a Two-component System
Infrared Spectroscopy
Infrared spectroscopy (IR) is one of the tools we will use to help identify or confirm the production of organic compounds along with melting points, boiling points, and gas chromatography. Spectroscopy is a very powerful analytical tool to all experimental science. Interestingly, one can learn a variety of different things about materials by matching the correct spectroscopic region, in terms of wavelength or frequency, with the particular problem being studied. The infrared spectrum of a compound can be used much like a fingerprint in determining its identity.
Radiation from different regions of the electromagnetic spectrum reacts with matter in different ways. In the infrared region (the wavelengths just longer than visible light) the primary interaction is between the infrared "photons" and the vibration of the chemical bonds. When the infrared photons have the right energy to cause "stretching" or "bending" of a particular covalent bond, the detector in the instrument receives less energy and an "absorption band" results. The "stretching bands", which occur at higher wavenumbers (the wavenumber is defined as the inverse of the wavelength in cm and has the units of cm-1), that are distinct for each type of covalent bond and, therefore, for each functional group. Thus, one can identify the presence (or absence) of a particular covalent bond (functional group) by the presence (or absence) of its characteristic absorption band. Some of these are given in the following table (Table 3).

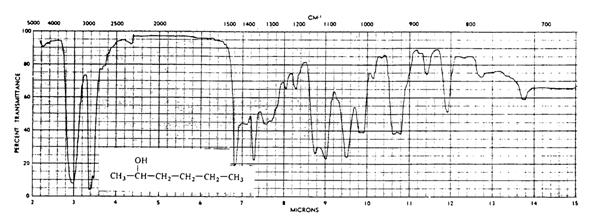
For example, we can identify functional groups in the above spectrum by using this table. This infrared spectrum has absorption bands at 3350 cm-1 (strong and broad) caused by O-H stretching of an alcohol, at 2960 cm-1 (strong) due to stretching of sp3 C-H, and at 1470 cm-1 (medium) caused by C-H bending of sp3 C-H. (Note the presence of these bonds in the formula of 2-hexanol). Comparison of the bonds present as shown by the infrared spectrum with the structural formulas of the possible or expected compounds will allow you to identify or confirm the identity of a compound.
Spectra of liquid samples with boiling points above 90°C can be obtained using two sodium chloride plates. A capillary film is formed between the two plates is formed by placing 1-2 drops on one plate and placing the other plate over this plate. The film thickness can be decreased by firmly pressing the plates together. Care should be taken to never touch or get water on the flat surface of sodium chloride plates, since water will cloud the surface of the plate. Hold the plates by their edges and keep your free hand under the plates so they don’t fall on the ground. Review the instructions posted by the IR (if available) and see you TA about obtaining a spectrum on the IR instrument. After obtaining the spectrum, slide the plates apart and clean them in the hood with acetone and store them in a desiccator. Be sure to hold both plates when transporting to avoid dropping a plate. IR spectra of liquids can also be taken using AgCl plates or a sealed cell for volatile liquids or solutions.
To take the IR spectrum of a solid, make a KBr pellet by grinding about 0.01 g of the solid compound in a use a clean and dry mortar and pestle for about 5 minutes. Mix this ground sample with about 0.5 g of KBr and grind until the mixture is a fine powder. Using a clean and dry metal pellet cell with one bolt screwed in half way, just cover the end of the bolt with the powder and tighten the other bolt into the nut using the vice and #14 wrench. Wait one minute and carefully unscrew the bolts. A slightly transparent pellet should remain in the cell. Add more powder if the pellet breaks or less if the pellet is opaque. Slip the nut holding your pellet into the white cell holder, place in the IR, and take the spectra. If the peaks are too short add more solid compound to the mixture or if too tall add more KBr and then repeat this procedure. IR spectra of solids can also be taken using the solution or mull methods.
IR spectra of both liquids and solids can also be run using attenuated total reflectance (ATR) FT-IR. For more information on this technique go to:
http://en.wikipedia.org/wiki/Attenuated_total_reflectance
This section has provided you with a brief introduction to infrared spectroscopy. You should now read the information on infrared spectroscopy in your lecture text. Try some of the problems involving IR spectra in the back of the chapter in your lecture text on infrared spectroscopy. For more information on FT-IR go to: http://infrared.als.lbl.gov/FTIRinfo.html